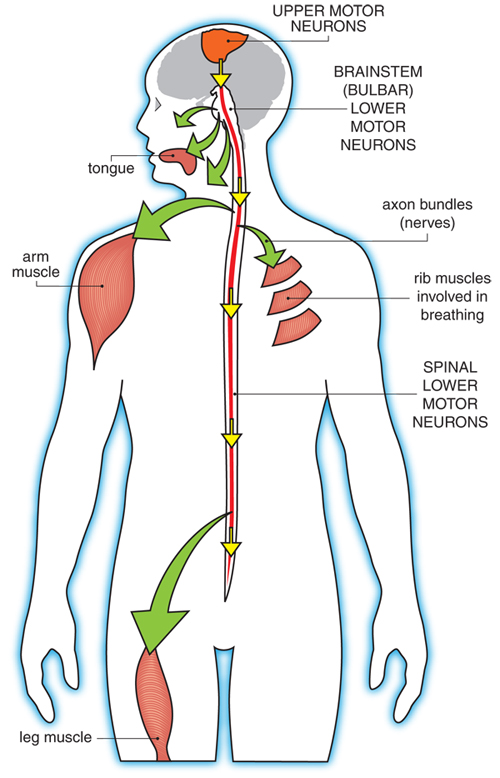
5 Nursing Diagnoses for ALS:
If you would like more information on the nursing care and management of patient’s with ALS. I would highly suggest going to this website, as it has a lot of useful information on how to care for these patients safely. It not only provides information on what to assess in these patients, but also how to manage these problems that pop up.
- Potential for injury related to impaired physical mobility
- Pt. has decreased muscle use and therefore they are at more risk for pressure ulcers and also injury from falling.
- Impaired urinary elimination related to progressive loss of mobility and dehydration
- Pt. has decreased swallowing and loss of mobility which leads to them being unable to drink a lot of fluids to produce enough urine, and also the loss of mobility prevents them from walking to the bathroom and being able to urinate by themselves properly. They may need help going to the bathroom as the disease progresses.
- Impaired nutritional status related to inability to swallow.
- Pt. has impaired muscle use of throat muscles, therefore they have decreasing ability to swallow and eat food properly, putting them at risk for impaired nutritional status as they are not getting enough of different types of foods as the disease progresses.
- Ineffective airway clearance related to impaired swallowing, gag reflex, and cough.
- Impaired muscles - abdominal, throat - lead to inability to cough and clear secretions in their throat.
- Impaired verbal communications related to altered volume of speech, altered clarity of speech and loss of speech.
- Muscles of speech (tongue muscles, mouth muscles, throat muscles) are affected by the degenerative disease, which decrease ability of the patient to use those muscles, which causes garbled speech.
|
Nursing Diagnoses
|
Goal
|
Intervention
|
Outcome
|
|
Potential for injured related to impaired physical mobility.
|
Promotion of activity and exercise.
Promotion of proper positioning to prevent decubitus ulcers.
|
ROM exercises to prevent contracture & pain in joints.
Change positions every 2 hours; after each change of position, check
for redness over bony prominences.
|
Patient will be active for as long as they can, and can remain more
comfortable so their joints and muscles die slower.
Patient will not get pressure ulcers, since they are immobile, by
turning them often you prevent skin breakdown.
|
If you would like more information on the nursing care and management of patient’s with ALS. I would highly suggest going to this website, as it has a lot of useful information on how to care for these patients safely. It not only provides information on what to assess in these patients, but also how to manage these problems that pop up.
Source:
Association, A. (2014). Nursing Management in ALS. Retrieved from ALS Association: http://www.alsa.org/als-care/resources/publications-videos/factsheets/nursing-management-in-als.html